Ecology, Evolution, and Bioinformatics
As a computational virologist, my research focuses on developing and applying computational methods to resolve complex microbial and viral communities from large-scale multi-omics data. Since my Ph.D. at Kyoto University in 2019, I have utilized metagenomics to decode the "dark matter" of our ecosystems, particularly focusing on the diversity and evolution of environmental viruses. By moving beyond traditional cultivation methods, computational biology allows us to uncover the hidden dynamics of the biosphere.
A central paradigm in my work is the genome-resolved approach. By reconstructing viral and microbial genomes directly from complex environmental samples, we can bypass the limitations of classical isolation. This computational approach fundamentally reshapes our understanding of biological diversity, revealing that extended environmental viruses possess complex genomes enriched with host-manipulation and metabolic genes. Such discoveries answer key questions about evolutionary relationships, functional potential, and how microbes influence global biogeochemical cycles.
However, recovering genomes is only the first step. To understand functional networks, I build analytical frameworks that integrate host datasets and phylogenetic inference to decode biological interactions. We still face fundamental questions: Who are they (Biodiversity)? Where are they (Biogeography)? When do they appear (Dynamics)? What do they infect (Network & Host Range)? How do they infect (Infection cycle)? Why do they evolve (Evolution)?
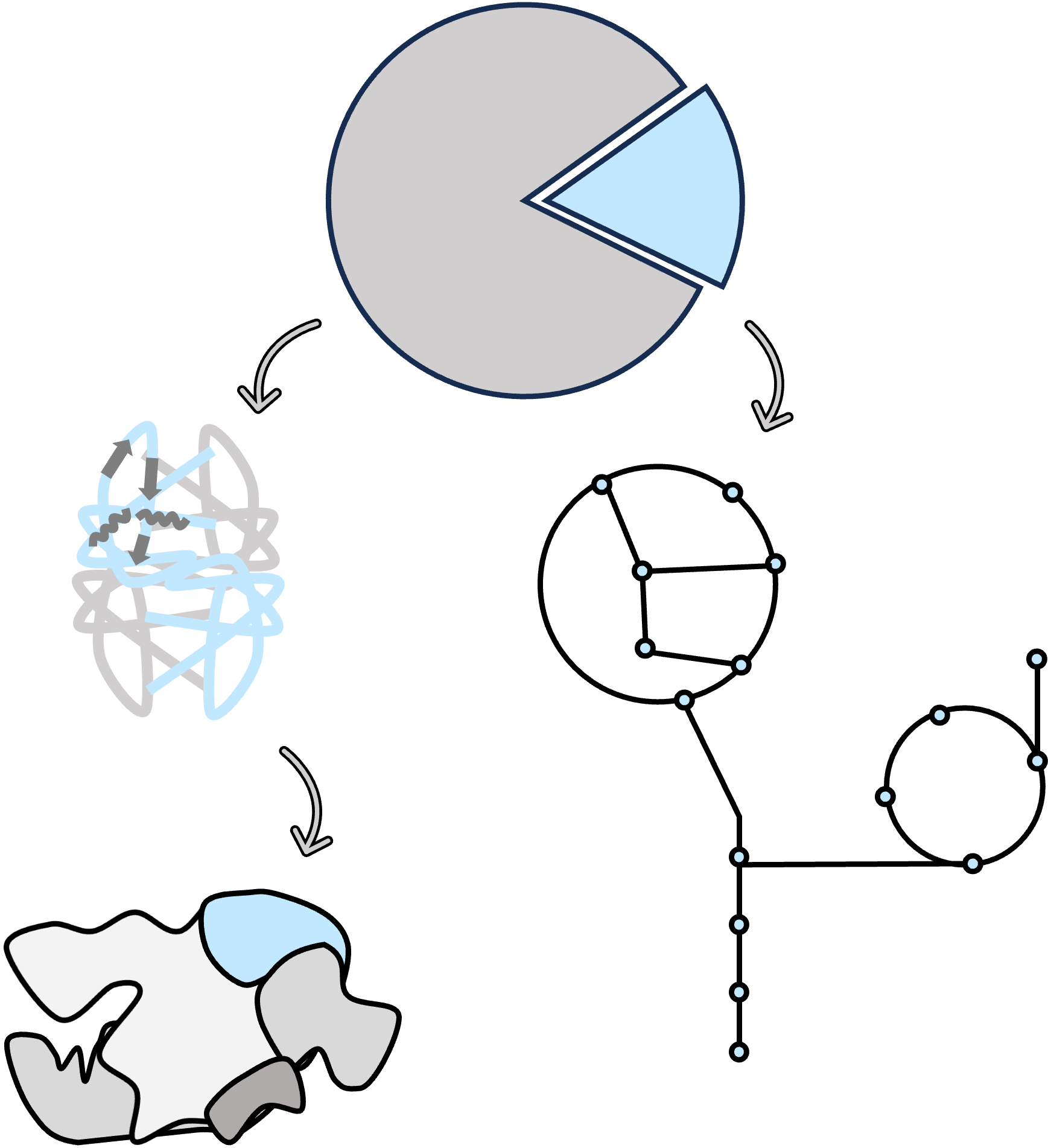
These multidimensional questions demand a data-driven approach. I aspire to push the boundaries of knowledge by organizing fragmented genomic sequences into an integrated, Computable Network of Life. To achieve this, I have structured my research program around four mutually interacting modules.
Diversity and evolution
Currently, large databases primarily include experimentally validated species. However, metagenomic analyses have revealed orders of magnitude more diversity. Defining microbial and viral diversity computationally is challenging because: 1) Target genomes are often highly complex, containing repetitive sequences and high intraspecies diversity; 2) Many lineages lack universal marker genes; 3) There are very few reference genomes mapped to isolated hosts.
In bioinformatics, our goal is to build robust analytical pipelines. An effective data-driven approach is to scan for highly conserved signature genes. By doing so, we computationally discovered a phylum-level new lineage related to herpesviruses, named "Mirusviricota" (Nature 2023). We also defined flexible core gene criteria that led to the identification of over 1000 new viral species from fragmented metagenomic assemblies (mSystems 2024). Beyond free-living virions, I systematically screen eukaryotic genomes to identify integrated viral signals, revealing massive 1.5 Mb endogenous viral regions (Virus Evolution 2023).
Why should we study this genomic diversity? Because genome signatures serve as digital "fossils" that guide evolutionary inference. Using robust phylogenetic methods, we untangle horizontal gene transfer (HGT) events, providing algorithmic evidence that herpesviruses and giant viruses share complex evolutionary trajectories (Nature 2023, MBE 2024). By integrating these evolutionary frameworks, we uncover how organisms co-adapt to harsh environmental and clinical stresses through genomic plasticity (Nature Communications 2023).
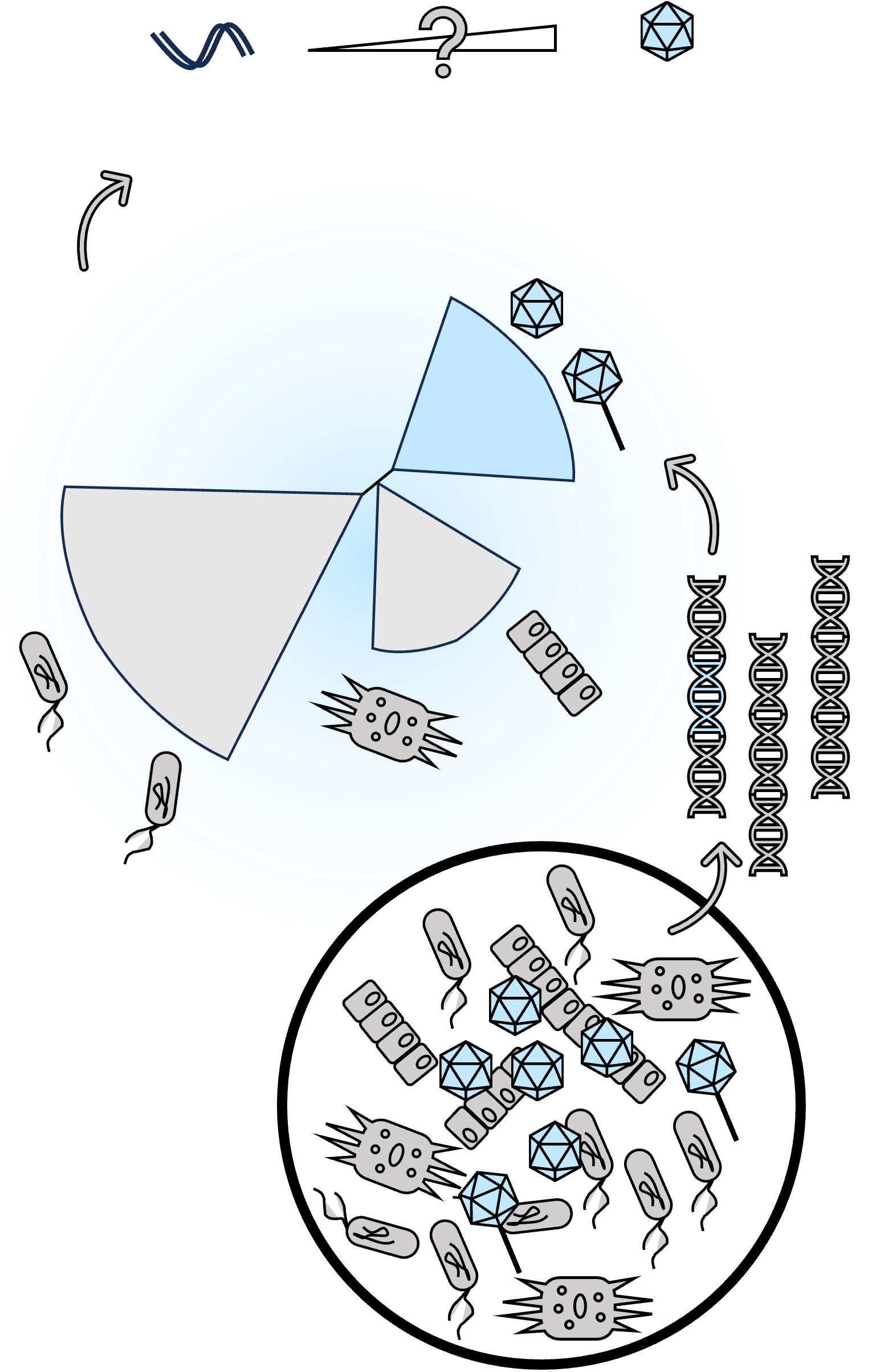
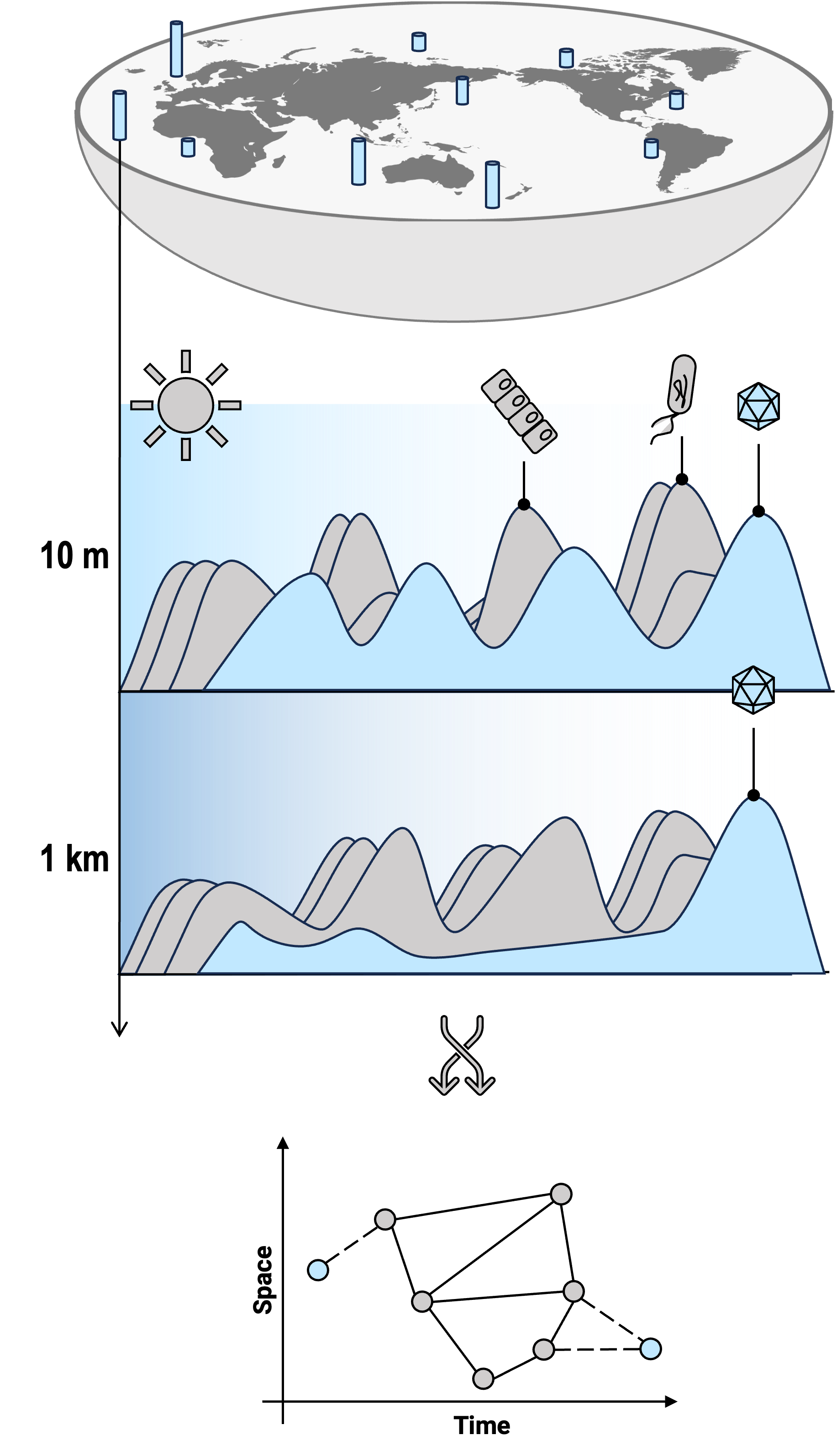
Giant viruses are widespread, abundant, and active in the ocean. Endemism was observed that a considerable proportion of unique populations have been found specifically in the Arctic Ocean (Nature Communications 2023). Besides the open ocean, giant viruses have also been detected in various aquatic environments, such as a eutrophic coastal inlet (mSystems 2024) and deep freshwater lakes (ISME J 2024). Giant viruses exhibit specific dark water temporal patterns. Overall, aquatic ecosystems harbor far more giant viruses than terrestrial or host-associated environments. However, a pithovirus lineage was discovered in numerous clinical patient samples across a three-year duration, and was also detected in widespread underground water samples (Coming Soon). In addition, other eukaryotic viruses, such as PLVs (Polinton-like viruses), are also abundant in aquatic ecosystems.
How are these viruses so successful in aquatic ecosystems? We observed a coastal giant virus community exhibits synchronous seasonal cycles with eukaryotes and year-round recurrence (mSystems 2024); nonetheless, most individual viral populations tend to be specialists rather than generalists. And we found that viruses with high microdiveristy tend to be generalists. So, by far I can gvie two strategies for marine eukaryotic viruses: first, by largely shifting their gene repertoires to adapt to changing hosts and ambient environments; and second, by accumulating sufficient mutations, which enhances their resilience and adaptability within complex ecosystems. Do I believe they are all the answer? Definately no.
Distribution of eukaryotic viruses
Host infection
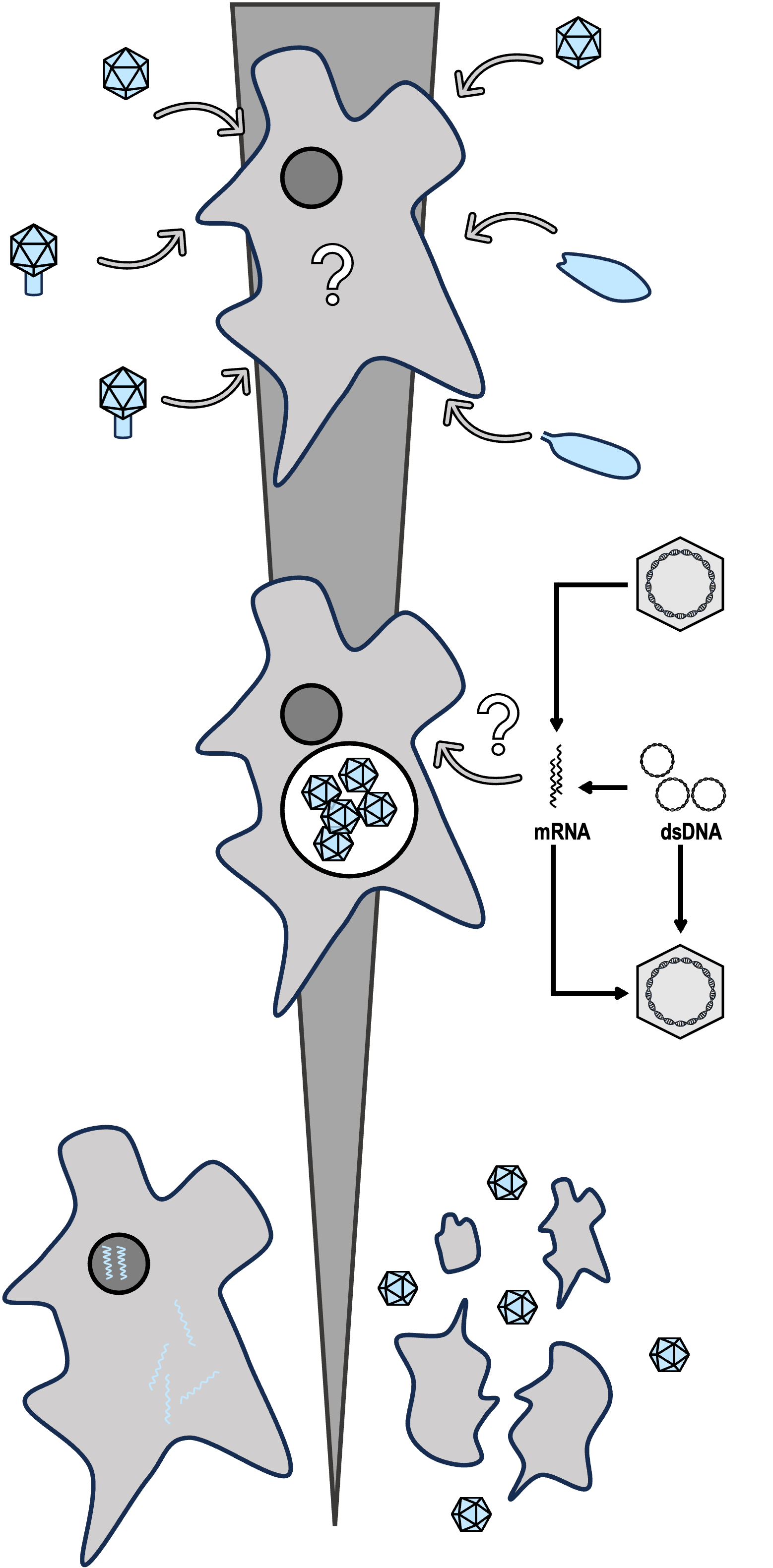
We are familiar with classic predator–prey dynamics in the macroworld. The microscopic world is equally fascinating, though relationships must often be inferred indirectly. By mapping complex ecological communities, we find unique life strategies ranging from "killing-the-winner" lytic pathways to persistent integration into host genomes. I hypothesize that horizontal interactions dynamically drive much of eukaryotic evolution (Communications Biology 2025).
To uncover these intricate relationships, my research heavily utilizes network inference and systems biology. One foundational aspect of my work is developing predictive bioinformatic methods. I engineered quantitative predictive models for host-virus interactions using co-occurrence networks and machine-learning-informed phylogenetic analysis (mSphere 2021). Additionally, approaches such as detecting HGT events (Nature 2023) and systematically screening for endogenous viral elements (Curr Biol 2024) provide rigid algorithmic evidence for mapping the global interactome.
Understanding viral functions is essential for revealing how viruses interact with their hosts, influence ecosystems and have evolved. Most of people believe that all viruses need to do is replicate themselves. So why do giant viruses being gigantism in their genome sizes and functions, such as Pandoraviruses, encode more than 2,000 genes? This seems counterintuitive given their reliance on host machinery. The presence of such large and complex genomes suggests there must be important trade-offs at related to host manipulation, autonomous functionality, or adaptation to diverse environments. Understanding these trade-offs is key to uncovering the evolutionary strategies and ecological roles of giant viruses.